KCC2 on Axotomized Motoneurons Is Regulated Differently Than in Systems Previously Described
Material below summarizes the article Removal of the Potassium Chloride Co-Transporter From the Somatodendritic Membrane of Axotomized Motoneurons Is Independent of BDNF/TrkB Signaling but Is Controlled by Neuromuscular Innervation, published on September 20, 2019, in eNeuro and authored by Erica Tracey Akhter, Ronald W. Griffith, Arthur W. English, and Francisco J. Alvarez.
Highlights
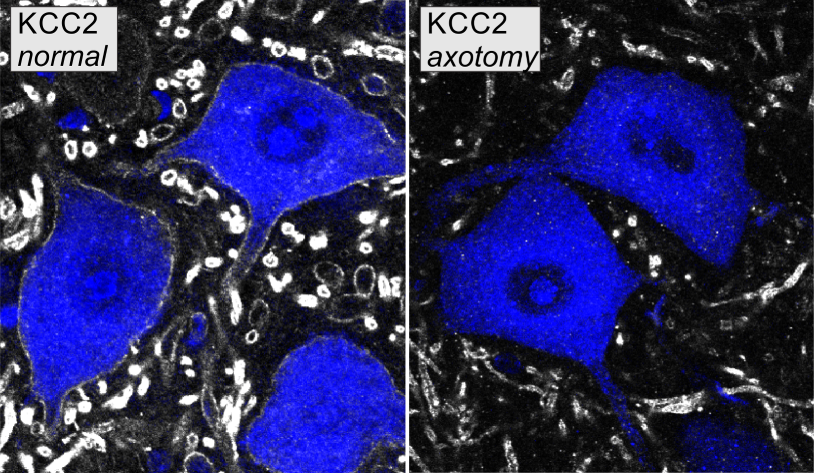
- Potassium chloride-cotransporter 2 (KCC2) mRNA and protein are downregulated within days of peripheral axotomy of spinal motoneurons.
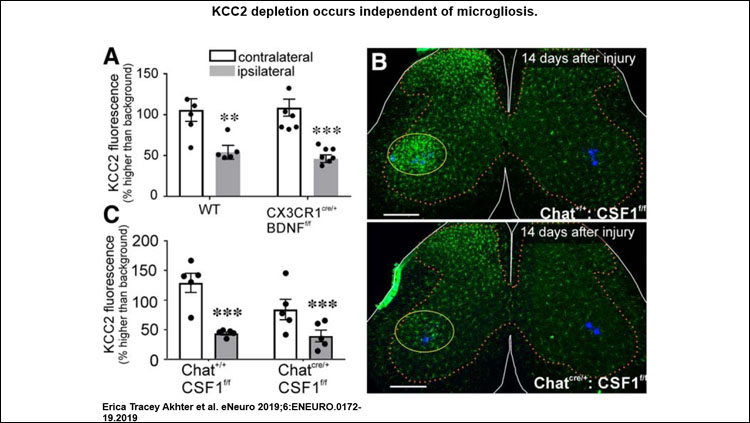
- KCC2 downregulation on axotomized motoneurons is not dependent on microgliosis or BDNF/TrkB signaling by difference to pathways reported previously in other disease and pathology associated with KCC2 dysregulation.
- Extensive KCC2 depletion is tied to the regenerative state and recovers after motor axons reinnervate muscle, suggesting that GABA/glycinergic synapse action in the absence of KCC2 might contribute to axon regeneration.
 |
|
|
Study Question
How is KCC2 downregulated in spinal motoneurons after nerve injury? The goals of the study were to elucidate the extent of KCC2 loss in cell body and dendrites and the dynamics and mechanisms of loss and recovery.
How This Research Advances What We Know
KCC2 regulates the chloride concentration within most neurons and therefore the strength and direction of GABA/glycine signaling. We know that KCC2 is depleted in a variety of injury states and diseases. KCC2 downregulation disinhibits the neurons from increasing their activity, and this has been associated with negative outcomes from motor spasticity to hyperalgesia or epilepsia.
Motoneuron activity is known, however, to help regeneration of their axon in injured nerves. We propose that KCC2 disappearance in motoneurons undergoing axon regeneration after nerve injuries may actually be helpful, but this question has never been investigated.
We found that the well-described BDNF/TrkB pathway that regulates KCC2 in many neurons after injury or pathology is not relevant in axotomized motoneurons. In contrast, KCC2 gene expression seems regulated by motor axon innervation of muscle. We suggest for the first time KCC2 gene regulation by synaptic targets.
Experimental Design or Methodology
We performed sciatic nerve injuries in mice and tracked KCC2 on cell bodies and dendrites of injured motoneurons using immunohistochemistry and RNAscope to respectively detect the protein or mRNA. Both males and females were used to ensure no potential sex-dependent effects were missed.
Prior to injury, we labeled the motoneurons from one specific muscle to investigate the same cell population in all animals. Labeling was accomplished with a long-lived retrograde tracer (Fast Blue) that fills the cell body or by AAV transfection with mCherry to fully label the dendrites. We analyzed motoneurons after injuries in which we allowed or prevented muscle re-innervation from occurring. KCC2 recovery with muscle re-innervation was correlated with neuromuscular re-occupancy by motor axons and motor end plates.
To analyze the BDNF/TrkB pathway, we used transgenic mouse models for cell-specific gene deletion of BDNF, TrkB chemogenetics, or ANA-12 TrkB pharmacology. All quantifications were done by blinded experimenters.
Results
Following sciatic nerve injury, KCC2 mRNA was downregulated in motoneurons within three days of axotomy. Two weeks following injury, KCC2 protein was consistently depleted on motoneuron somata and throughout much of the dendritic arbor. The losses on KCC2 mRNA and protein were almost complete, and their time courses suggest that the loss of protein is a consequence of KCC2 gene expression downregulation, not of membrane trafficking.
KCC2 depletion was sustained for two months after injury when regeneration was prevented. KCC2 was restored, however, when the sciatic nerve was repaired to allow regeneration and muscle reinnervation. The level of KCC2 restoration was positively correlated with the degree of neuromuscular junction reinnervation, suggesting that signals from muscle, informing about re-innervation, regulate KCC2 expression.
KCC2 depletion occurred independently of mechanisms previously established for other regions of the spinal cord after peripheral nerve injury (PNI). It was not dependent on microgliosis, BDNF release from microglia or motoneurons, or TrkB receptor signaling. Thus, none of the mechanisms usually reported to deplete KCC2 in central neurons after various pathologies were significant for KCC2 regulation in motoneurons after axotomy. KCC2 expression in axotomized motoneurons may be regulated by yet-unidentified retrograde signals from muscle.
Interpretation
The extent of KCC2 loss exceeds what is reported in other pathologies. This massive loss should not be interpreted as “disinhibition” because of reduced GABA/glycine synapse efficacy, but as a change in the driving force sign.
Similar to what was reported in brainstem motoneurons, GABA/glycinergic activity likely depolarizes axotomized spinal motoneurons. This challenges common ideas about GABA/glycine synapses helping maintain injured motoneurons silenced to focus on regeneration, rather than firing. On the contrary, GABA/glycine synapses are likely activity drivers after axotomy.
Muscle innervation regulates KCC2 expression in motoneurons, providing a convenient on/off switch for regenerating motoneurons. This is different from the BDNF/TrkB dependent mechanism of KCC2 regulation in spinal neurons transmitting pain after PNI.
While surprising, the difference is reassuring from a clinical perspective. Treatments to treat neuropathic pain by preserving KCC2 activity after PNI can be dissociated from mechanisms of KCC2 downregulation in motoneurons, maintaining any beneficial actions on regeneration.
Visit eNeuro to read the original article and explore other content. Read other summaries of eNeuro and JNeurosci papers in the Neuronline collection SfN Journals: Research Article Summaries.
Removal of the Potassium Chloride Co-Transporter From the Somatodendritic Membrane of Axotomized Motoneurons Is Independent of BDNF/TrkB Signaling but Is Controlled by Neuromuscular Innervation. Erica Tracey Akhter, Ronald W. Griffith, Arthur W. English, and Francisco J. Alvarez. eNeuro 20 September 2019, 6 (5) ENEURO.0172-19.2019; DOI: 10.1523/ENEURO.0172-19.2019
About the Contributors





